让Claude成为化学家
Making Claude a chemist
Anthropic 发布白皮书,评估 Claude 模型在化学 NMR 谱图分析中的表现。研究选取 20 种化合物,对比 Opus 4.7、Opus 4.6、Sonnet 4.6 与 ChemDraw、MestReNova 在正向预测(谱图模拟)和反向解析(从谱图推导结构)上的性能。Opus 4.7 在氢谱预测中平均误差为 ±0.079 ppm,碳谱与 MestReNova 持平(±1.37 vs ±1.48 ppm),并在峰形匹配和子峰间距预测上优于专用软件。在反向任务中,Opus 4.7 成功恢复全部 8 个简单结构和 4 个复杂结构(共 7 个)。该研究由 Anthropic 化学家 David Kamber 主导,旨在推动 AI 辅助化学日常分析工作。
摘要:我们正与世界一流的合成化学家、计算化学家和分析化学家合作,让 Claude 在化学领域表现更出色。本文分享了这项工作的首个成果,Anthropic 化学家 David Kamber 考察了 Claude 如何处理化学家最常用的分析输入——NMR 谱图。 在处理分子时,化学家需要在白板上的手绘结构、仪器读数、数据库查询字符串以及专利和出版物中的技术符号之间切换。每种表示方式都编码了相同的底层化学信息,但各自需要不同的熟练度。例如,咖啡因的草图能让化学家发现它与腺苷(人体困倦信号)的相似性,并预测它通过阻断受体来保持我们清醒。然而,同样的草图无法帮助化学家将其与其他外观几乎相同的分子区分开来。
了解化学家正在处理哪种分子至关重要。化学支撑着我们摄入的食物和药物,以及我们的乳液、油漆和塑料。在相同原子之间重新排列少数化学键,葡萄糖就会变成果糖——这两种分子共享同一分子式,但通过完全不同的代谢途径处理。将分子翻转成其镜像,镇静剂就会变成致畸剂,正如沙利度胺灾难中所发生的那样。¹ 化学家的日常工作依赖于在适合特定任务的任何表示方式中正确解读这些信号。
在这些表示方式之间进行转换(从图中追踪结构、将仪器读数与预期产物进行比对、以正确符号查询数据库)既耗时又难以大规模跟上——CAS(最大的化学注册数据库)已收录超过 2.9 亿种已公开物质,并且每天新增约 15,000 种。
AI 完全有能力承担这一研究负担,但在化学领域,它仍然在很大程度上停留在理想层面。多年来,机器学习工具一直被定位为逆合成分析(从目标分子反向推导到更简单前体以规划合成路线)、反应预测和性质估计的变革性工具,但这些工具所需的数据一直难以获得——零结果数据稀疏、格式不一致,并且被锁定在订阅期刊的付费墙后(以及非结构化的支持信息中)。逆合成分析就是一个典型例子——功能强大的 AI 工具已存在多年,但采用率参差不齐,普通学术或小型实验室的化学家仍然不使用它们。
即便如此,AI 的进步终于触及化学领域。当今的前沿模型是多模态的,并且具备显式推理能力。它们可以直接从期刊图或手绘草图中读取化学结构,而无需依赖预先整理的分子数据库。它们还能以实际发表的形式读取方法部分或支持信息中的实验细节。它们还可以逐步展示推理过程,这意味着化学家可以审核输出结果。这些都不能消除该领域多年来一直描述的数据问题,但它改变了哪些问题在此情况下仍然可以处理。
最终,我们的主张是适度的:Claude 开始有意义地协助化学家进行日常的转换、回忆和整合工作,以补充他们的判断力,我们计划继续扩展其有用性。今天我们发布了加速这项工作的第一份白皮书。它处理的是化学家最常用的分析输入:NMR 谱图。
Claude 与 ChemDraw 在 NMR 预测和结构解析上的对比
完整版本可在此处找到:链接
几乎所有小分子——药物、农药、染料、香料、聚合物、DNA 或蛋白质亚基,以及功能性无机或固态材料——的存在都源于化学家确定了其结构。鉴于这些分子无法用显微镜观察,化学家必须依赖光谱分析,用光、无线电波或磁场探测分子。给定分子吸收、发射或偏转这种能量的方式为化学家提供了一种模式,即谱图,他们可以据此解析其结构。
NMR 波谱学——化学家依赖的标准技术之一——是合成化学中最耗时的步骤之一;对于每种化合物,化学家必须手动将谱图中的每个峰与所提出结构中的原子进行匹配。在这份白皮书中,我们测试了 Claude 与化学家目前依赖的专用 NMR 软件相比表现如何。我们测量了三个 Claude 模型(Opus 4.7、Opus 4.6、Sonnet 4.6)与 ChemDraw 和 MestReNova 在 20 种化合物上的表现,这些化合物选自模型训练截止日期后发表的合成化学预印本,以避免选择偏差。ChemDraw 和 MestReNova 都进行正向预测,使用绘制结构来模拟将产生的 NMR 谱图。除了正向预测,我们还希望看看 Claude 能否反向进行——从实验谱图出发,提出其背后的结构。这是更困难的任务,也是现有软件目前留给化学家的工作。
为了设置评估,我们从模型训练截止日期后发布的 ChemRxiv 预印本² 中选取了 20 种化合物,从每篇论文中提取了第一个完全表征的新分子。这 20 种化合物涵盖四个结构家族,每个家族五种化合物,每个家族的选择是因为它涉及不同类型的 NMR 挑战。每个工具都获得了以 SMILES 字符串(化学家用来将分子输入软件的行文本符号)编码的结构,并被要求预测每个氢和碳峰在 1D NMR 谱图(以 ppm 为单位测量化学位移的水平轴)上的位置。鉴于 NMR 样品溶解在液体中,并且溶剂(氯仿、DMSO 等)的选择会略微移动峰位置,每个工具被告知要根据已发表论文中化学家使用的溶剂来预测谱图。
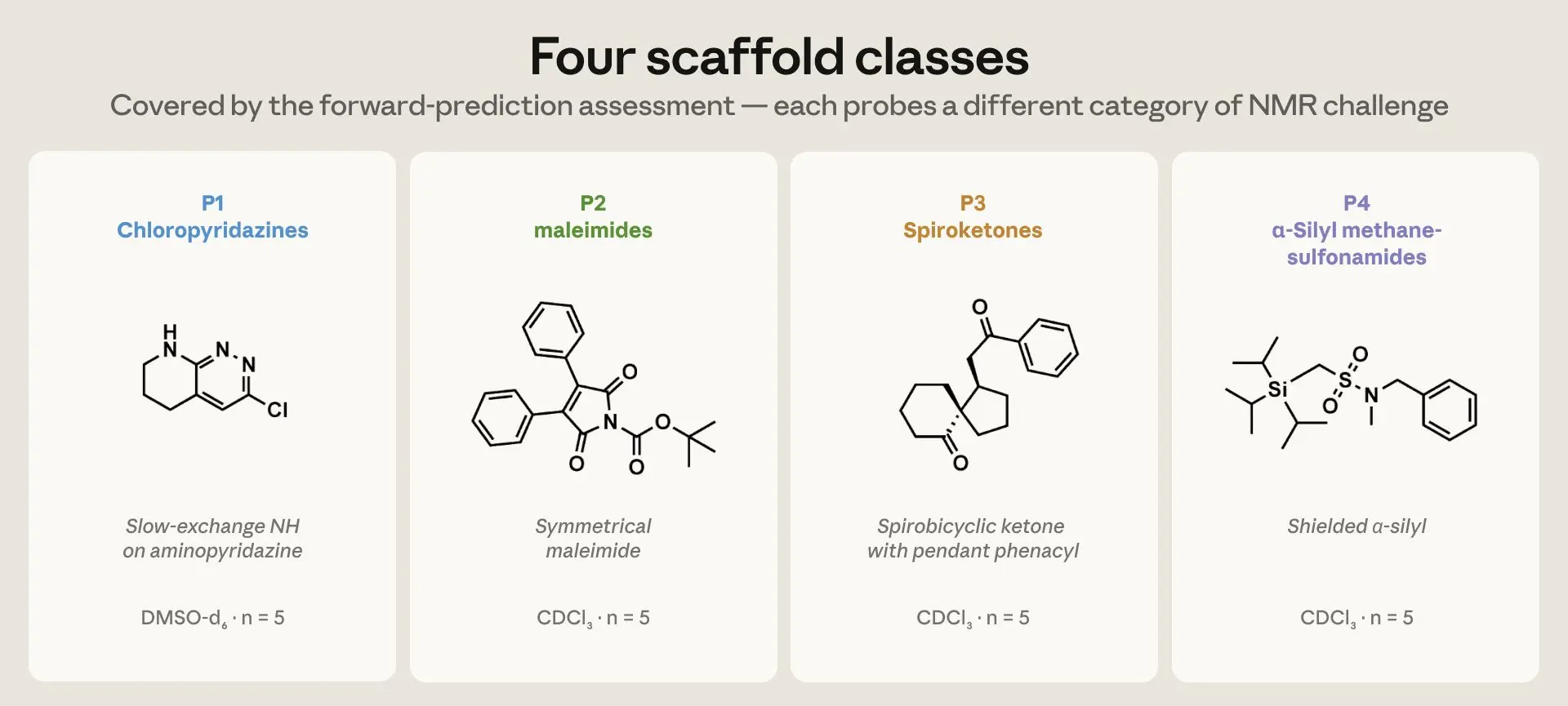
图 1. 正向预测评估涵盖的四种骨架类别。每种都涉及不同类型的 NMR 挑战。P1 氯哒嗪在 DMSO-d₆ 中具有氨基哒嗪上的慢交换 NH;P2 Boc-N-芳基马来酰亚胺和 N-Boc 炔酰胺涉及 α-乙烯基-酰亚胺羰基和罕见的炔酰胺 α/β-碳对;P3 螺酮是具有苯甲酰甲基或乙酰基侧链和非对映异位 CH₂ 的螺双环酮;P4 α-硅基甲磺酰胺具有屏蔽的硅-α 碳。每类五种化合物,总计 n = 20。
由于语言模型的输出在不同运行之间会变化,每个 Claude 模型对每种化合物查询三次并取平均值;ChemDraw 和 MestReNova 每次返回相同答案,因此运行一次。然后我们将每个预测峰与其实验对应峰配对,并测量 ppm 差距。这些差距落在化学家认为正确的窗口内——氢为 ±0.20 ppm,碳为 ±1.0 ppm。
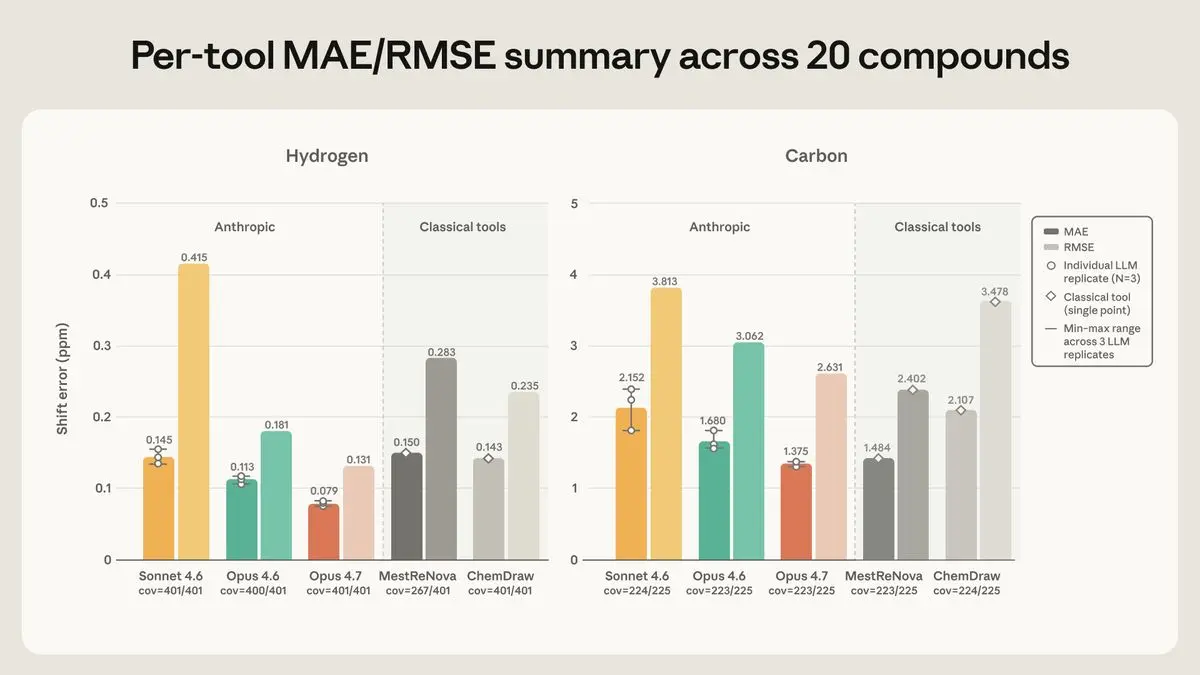
图 2. 20 种化合物正向预测的 ¹H(左)和 ¹³C(右)位移误差的各工具 MAE(深色阴影)和 RMSE(浅色阴影),每个工具下方显示覆盖率。Claude 柱状图:三次重复的平均值,附有最小-最大范围和叠加的重复数据点。经典工具:单点预测(无范围)。
在氢方面,Opus 4.7 最准确,平均误差为 ±0.079 ppm——远低于容差窗口的一半——并且落在窗口内的峰比例最高。在碳方面,Opus 4.7 和 MestReNova 基本持平,分别为 ±1.37 和 ±1.48 ppm;其余工具在两种元素上保持相同的排名顺序。Opus 4.6 不出所料地处于中等水平,Sonnet 4.6 最弱。它们之间的差距在一个众所周知的困难氢上最为明显——氯哒嗪家族中的一个 NH 质子,其真实位置落在 6.8 到 7.9 ppm 的狭窄范围内。Opus 4.7 将其放置得略低但一致;Opus 4.6 的猜测分散在几个 ppm 之间;Sonnet 4.6 将其放在 10–13 范围内,远高于其实际出现位置。
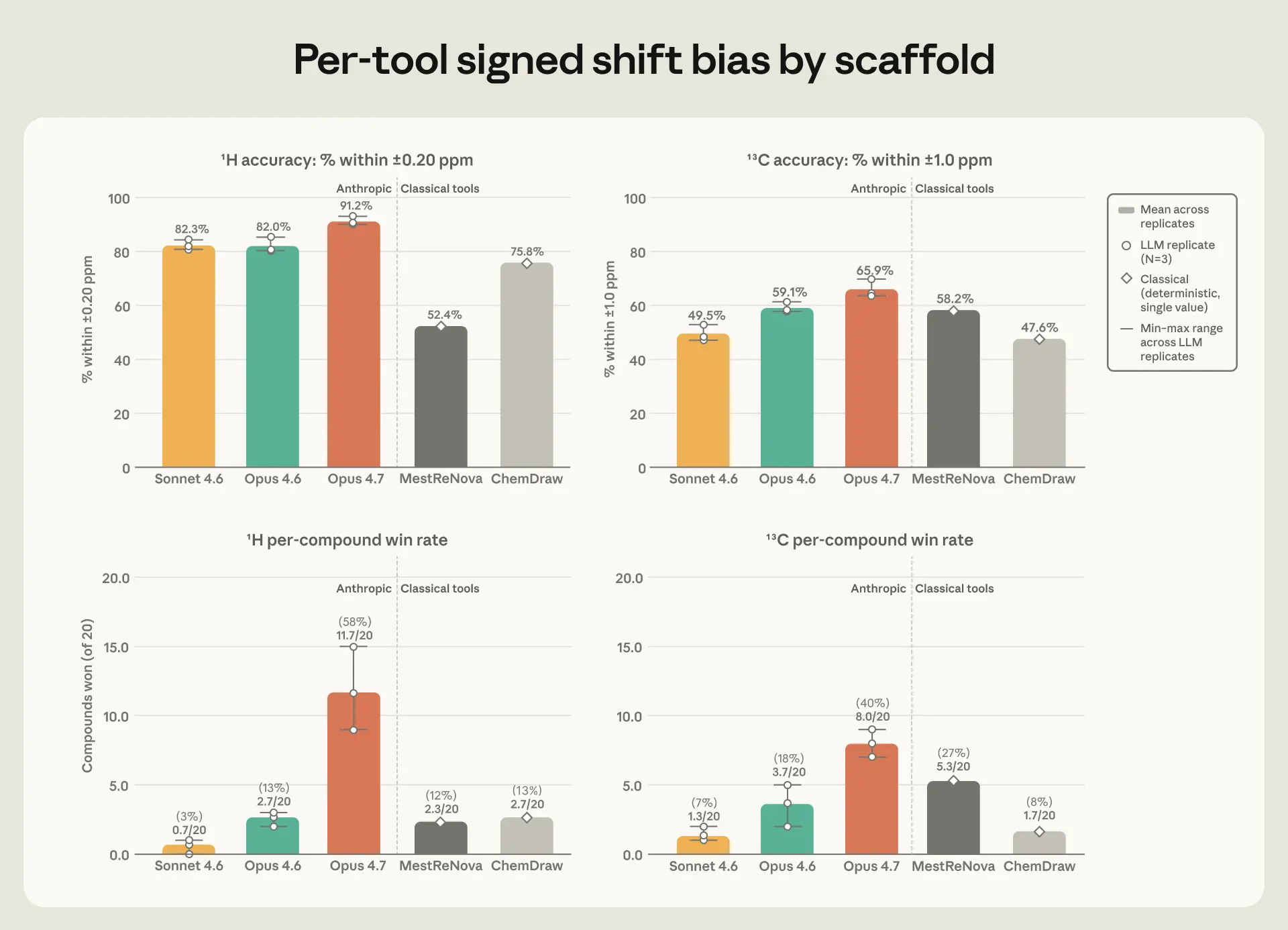
图 3. 顶部:实验原子在 ±0.20 ppm(¹H,左)和 ±1.0 ppm(¹³C,右)内的百分比。底部:每种化合物的胜率(工具在 20 种化合物中具有最低每种化合物 MAE 的化合物)。Claude 柱状图:三次重复的平均值,附有最小-最大范围;经典工具:单点预测。
虽然 Opus 4.7 的表现与 ChemDraw 和 MestReNova 相当,但在预测氢 NMR 峰的形状以及峰间距方面差距更大,这些特征也包含化学家与位置一起读取的结构信息。Opus 4.7 比任何其他工具都更频繁地匹配实验报告的分裂模式,并且所有三个 Claude 模型在大约 80% 的情况下预测了子峰间距在 0.5 Hz 以内——而 ChemDraw 和 MestReNova 为 26% 到 35%。Opus 4.7 在其三次重复运行中也最为一致:其平均误差在不同运行之间的变化小于其与次优工具之间的差距。
从那里,我们评估了反向预测(结构解析):我们能否从谱图中确定分子的结构?我们给 Opus 4.7 提供了 15 个解析问题,并让它每次提出最多三个排序的候选结构,重复三次。每个问题提供了化合物的精确分子式(来自高分辨质谱)及其氢和碳 NMR 谱图。这 15 个问题按难度划分。八个较简单的目标——单环或双片段分子——仅提供分子式和谱图。七个较密集的目标——稠环、螺环等——附带一个额外提示:反应中使用的起始原料的结构。
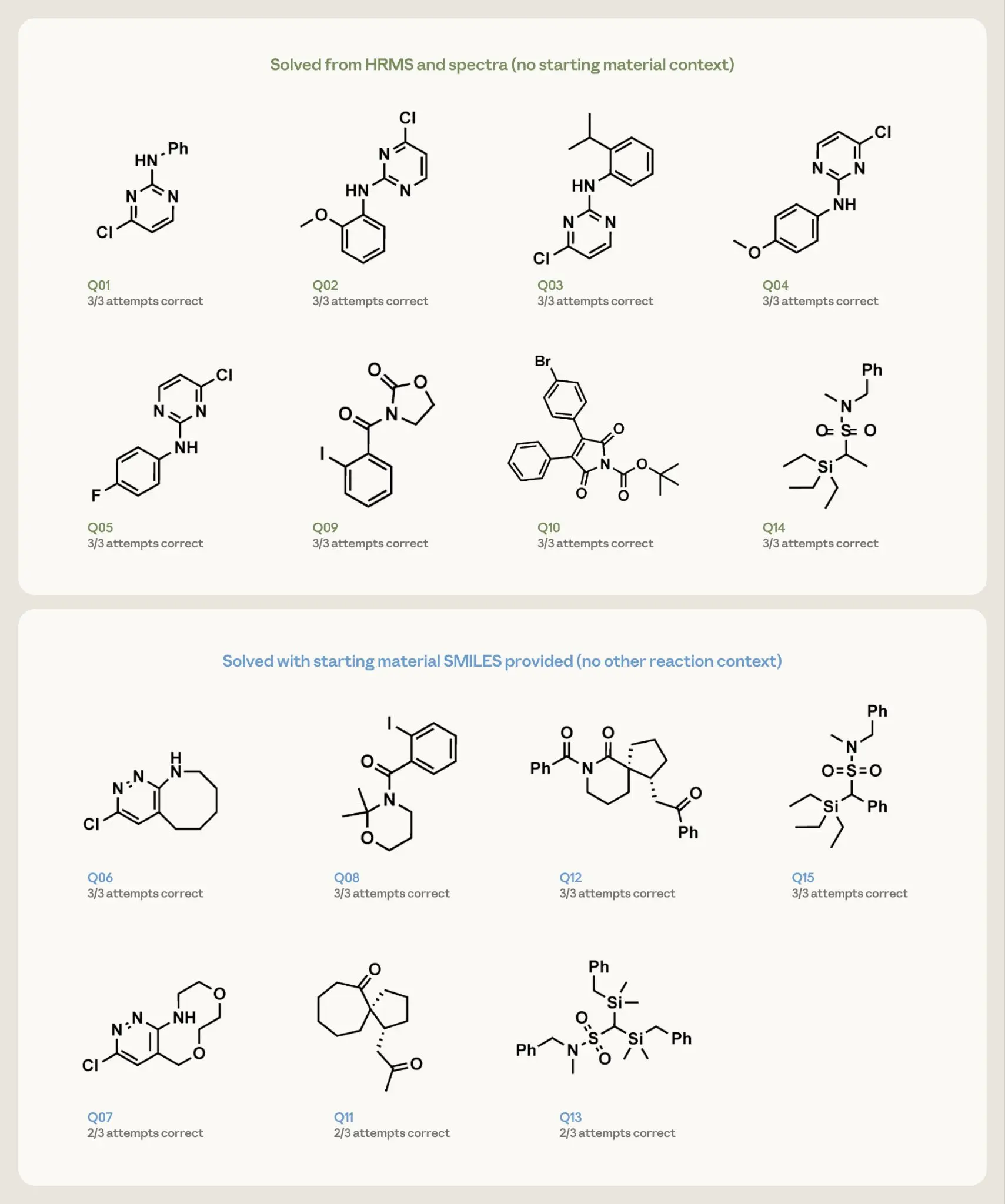
图 4. 15 个反向任务问题的结构解析结果。每个面板显示已发布的目标及其在 3 次尝试中的成功次数。边框颜色表示提示条件:绿色表示仅谱图和 HRMS,无起始原料背景;蓝色表示谱图、HRMS 和起始原料 SMILES,无其他反应背景。
Opus 4.7 仅凭谱图和分子式就成功恢复了所有八个较简单的结构,每次尝试都成功。在七个较困难的目标上,给定起始原料提示,它在所有三次运行中为其中四个返回了正确结构,在剩余目标的三次运行中有两次返回了正确结构。
最终,我们发现对于常规数据预测,Opus 4.7——一个没有化学特定微调的通用模型——现在平均而言与 ChemDraw 和 MestReNova 一样好或更好。此外,Claude 还可以反向处理问题,仅从 NMR 数据提出结构。专用的结构解析软件已经存在了几十年,但它通常需要 2D NMR(具有两个轴的谱图,输出是等高线图而不是一排峰)、专门培训和许可工具。Claude 使用化学家会粘贴到聊天中的相同高分辨质谱和 1D 峰列表即可完成,无需任何设置。
局限性
这项评估表明,通用模型可以与 NMR 软件竞争,甚至使 1D 反向解析变得可行。但存在一些值得注意的局限性。
- 首先,评估规模较小——正向任务涉及四个骨架的 20 种化合物,反向任务涉及 15 种——并且每种骨架贡献了一类故障模式。因此,模型性能应被视为指示性的而非精确的。
- 其次,对于最密集的反向目标,如果没有起始原料作为额外输入,模型可能会在推理中循环,而不承诺最终结构;这就是为什么七个较困难的问题是在提供起始原料结构而非仅谱图的情况下提出的。
- 第三,一些化学骨架未经过测试。例如,慢交换 NH 杂芳烃(其 N–H 与溶剂交换足够慢以留下尖锐 NMR 峰的芳香环)仅通过氯哒嗪进行采样,排除了相关系统(羟基吡啶、氨基噻唑和其他 DMSO-d₆ NH 活性骨架)。
- 第四,2D 实验(COSY、HSQC、HMBC)和立体化学按设计不在范围内,因为仅凭 1D NMR 无法确定构型。因此,复杂的天然产物化合物未进行评估。
- 最后,我们的溶剂覆盖范围仅限于 DMSO-d₆、CDCl₃ 和 D₂O,因此未评估甲醇-d₄、苯-d₆ 和丙酮-d₆。
理想情况下,我们希望看到这些数字在跨越 20–30 个骨架类别的数百种化合物上如何保持,每类至少 15 种化合物,以便将类内方差与工具间差异分开。我们还将评估氯哒嗪以外的 NH 活性杂芳烃,评估未测试的溶剂,并进行利用 2D 实验的两种任务版本。
展望未来
随着我们继续提高 Claude 在化学领域的表现,我们特别关注几个最拖慢化学家速度的瓶颈。
- 读取和渲染化学结构——将来自图、专利、幻灯片或草图的绘图转换为机器可读形式,并在结构表示与化学文献中使用的系统名称之间进行转换。
- 反应和合成推理——提出、评估和批评合成路线,预测结果,并思考选择性、条件和可能的副产物。
- 机理——以化学家实际使用的语言解释和测试反应机理,包括电子箭头、中间体和过渡态论证。
- 化学文献理解——阅读已发表作品中的化学内容,其中同一分子可能被绘制、命名、缩写或通过代码引用,并从方法部分、支持信息和专利中提取重要的化学信息。
这些并不都处于相同的成熟曲线上。在光谱分析已经足够成熟可以进行基准测试的地方,其他方面,如逆合成规划,仍在界定范围。随着我们对这些瓶颈有了更好的理解,我们将分享当前模型在哪些方面表现出色,以及它们仍然在哪些方面存在不足。我们的最终目标是确保在职化学家知道 Claude 在哪些方面可以节省他们的时间,以及他们仍然需要在哪些方面依赖自己的专业知识。
与我们合作
我们正在扩展 AI for Science 项目,以更明确地支持化学研究。如果您是一位研究人员,正在研究 Claude 可能有助于解决的问题,尤其是涉及我们描述的那种多模态推理的问题,我们期待您通过 scienceblog@anthropic.com 或通过 AI for Science 申请与我们联系。
脚注
- 一起事件,其中一种晨吐药物与全球超过 10,000 名儿童的严重出生缺陷有关。
- 我们从中提取化合物的四篇预印本:https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002274/v1,https://chemrxiv.org/doi/full/10.26434/chemrxiv-2025-59lfh,https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002423/v1,https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002316/v1。
相关内容
社会科学中的编码智能体
一项关于 1,260 名社会科学家对 AI 和编码智能体使用的调查结果。
Project Glasswing:初步更新
关于我们从 Project Glasswing 中学到的东西的早期更新。
2028:全球 AI 领导力的两种情景
我们对中美 AI 竞争的看法。